
Crouzon syndrome: an experience of surgical intervention at Bangabandhu Sheikh Mujib Medical University, Bangladesh
 0
0Abstract
Crouzon syndrome, also called craniofacial dysostosis is a rare autosomal dominant disease. It has a prevalence of 1 in 25,000 live births and it constitutes 4.8% of all craniosynostosis. Craniosynostosis, shallow orbits, maxillary hypoplasia, ocular proptosis and hypertelorism are the cardinal features of Crouzon syndrome. Here, the authors report a case of this rare entity who presented with brachycephaly, maxillary hypoplasia, shallow orbit, hypertelorism and bilateral proptosis. This patient’s overall management was done with the involvement of a multidisciplinary team.
Keywords
Introduction
Crouzon syndrome is an autosomal dominant disease which is also known as “branchial arch syndrome”. The first branchial (or pharyngeal) arch which plays a pivotal role in the formation of maxilla and mandible is affected in this syndrome. Disturbances in branchial arch development create widespread effects. Crouzon,[1] a French physician was the first to describe this syndrome. The most important characteristic of Crouzon syndrome is craniosynostosis. It usually presents as brachycephaly resulting in the appearance of a short and broad head. Hypertelorism, exophthalmos and beak-like nose are characteristic features. External strabismus is also a common feature. Lastly, hypoplastic maxilla results in relative mandibular prognathism and gives the impression of a concave face. Patent ductus arteriosus and aortic coarctation are also sometimes associated with Crouzon syndrome.[2]
Mutations of the fibroblast growth factor receptor 2 (FGFR2) and FGFR3 are the causative factors in the autosomal dominant Crouzon syndrome.[3]FGFR2 is the most commonly mutated gene, a missense at cystine 342 in exon 9.[3] Recent advances in our understanding of human genetic disease has led to a better understanding of how the particular gene perturbations produce cranial skeletal malformations.[4] Incidence of Crouzon syndrome is approximately 1 in 25,000 live births worldwide.[5] This syndrome makes up approximately 4.8% of all cases of craniosynostosis.[6] The differential diagnosis of Crouzon syndrome are simple craniosynostosis, Apert syndrome, Carpenters syndrome, Saethre-Chotzen syndrome and Pfeiffer syndrome.[7] Mutations of the FGFR3 gene in these patients may also be associated with acanthoses nigricans.[8]
Every child is different and their clinical manifestation depends on which sutures are fused first. The manage-ment is also based on the affected sutures. Surgery prevents the early closing of the suture lines of the skull and helps in the brain development. Blindness and mental retardation are the usual outcome if timely surgery is not performed.[9]
Case report
A 6-month-old female child came from Satkhira was admitted to Bangabandhu Sheikh Mujib Medical University presented with abnormal shape of the skull and markedly protruding eyes soon after birth. The baby’s head was disproportionately wide compared to its length, the back of the head was flat and it was wide and short [Figure 1]. She had visual problems due to shallow eye sockets and strabismus. For these complaints, she visited local doctors and was referred to this tertiary center for further evaluation and management. The prenatal, natal and postnatal histories were not significant. She is the second child of apparently healthy parents of consanguineous marriage. Other siblings or parents did not have a similar abnormality. Her developmental milestones were found to be normal. On developmental assessment, she can sit without support, transfers object and two-way vocalization were present. Her hearing was also intact.
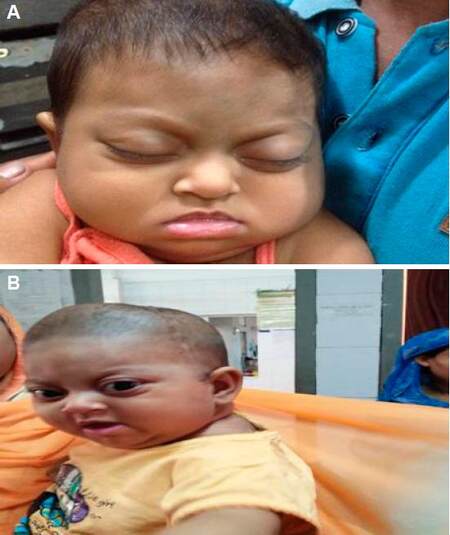
Figure 1. Comparison of (A) pre-operative and (B) post-operative (3 weeks later) of Crouzon syndrome patient
She had enlarged cranial vault with frontal bossing, maxillary hypoplasia and a relative mandibular prognathism was found. Ocular manifestations such as shallow orbits, hypertelorism, bilateral proptosis, exophthalmos and strabismus (non-paralytic) were present. Other facial features included short and incompetent upper lip, high arched palate, depressed nasal bridge and low set ears [Figure 1].
Audiological assessment was done. Hearing was found to be normal. Her hands and feet showed no obvious abnormalities. On anthropometry, her weight for age was just below the 10th percentile, length for age was at the 50th percentile, weight for length just below the 10th percentile. Other systems such as cardiovascular, gastrointestinal tract and genitourinary system were all normal.
On ophthalmological evaluation by fundoscopy, papilloedema was present on both eyes which correspond to grade 2 Modified Frisén Scale. The skull radiographs [Figure 2] revealed a “brachycephalic” skull shape, hypoplastic maxilla and a zygoma with shallow orbits. Other routine haematological and biochemical tests were within normal limits.
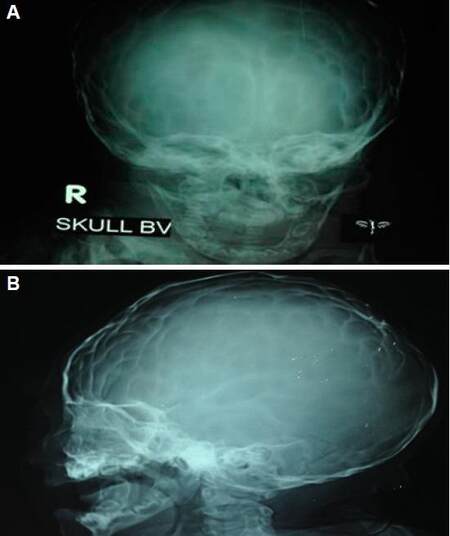
Figure 2. Copper beaten appearance, (A) flattening of frontal bone and prominent occipital bone, (B) fusion of coronal and sagittal suture of a 6 months old female baby
We counseled the parents about the nature and future of the disease. The baby was on complementary feeding along with breast feeding. Consultation from the eye department was obtained taken and they advised expectant treatment. Our treatment goal was to allow the increase in size of the skull to make room for the growing brain and to restore a normal head shape. So, we took advice from the neurosurgery department regarding the possibility of surgical intervention. Strip craniotomy was done by Pi procedure [Figure 3].
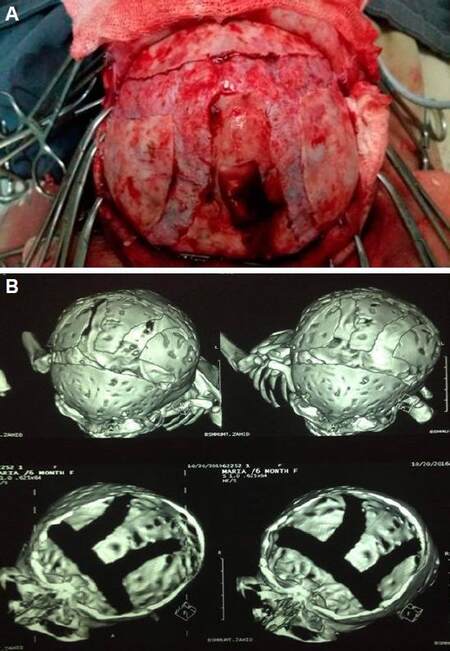
Figure 3. (A) Pre-operative view and (B) post-operative computed tomography scan showing Pi (π) procedure done in a 6 months old female baby
A section of skull, shaped like the Greek letter pi was removed. After surgery, an I/V antibiotic was given and there were no complications. As the condition of the baby was stable we discharged the baby and advised follow up. Patient came for follow up 21 days (3 weeks) later. Patient’s condition was stable, wound healed well, no neurological deficits were found. Fundoscopic examination showed that the papilloedema reduced from grade 2 to grade 1 based on the Modified Frisén Scale, although there was little change in exophthalmos and strabismus.
Discussion
Crouzon syndrome is one type of craniofacial dysostosis which is caused by premature obliteration and ossification of two or more suture lines, mostly coronal and sagittal sutures. Initially it was described as a hereditary syndrome of craniofacial synostosis, that includes a triad of skull deformities, facial abnormalities and exophthalmos. All these characteristic features were seen in our patient. The overall appearance of an infant with Crouzon syndrome can vary from a mild presentation with subtle midface deficiency to severe forms of multiple cranial sutures fused and marked facial and eye problems.[9] In the present case, premature closure of the sutures had caused restricted skull growth and lack of space for the growing brain resulting in compensatory change in the growth of skull and brain toward the frontal region.[9]
Presence of upper airway obstruction, respiratory distress and mental retardation is rare in these children.[9,10] Our patient was also developmentally age appropriate and there were no signs of respiratory difficulty.
Increased intracranial pressure may lead to optic atrophy, which can produce blindness if left untreated. Optic atrophy is frequently seen and has been reported in 30-80% patients. Affected individuals exhibit ocular malformations including hypertelorism and proptosis due to shallow orbits.[9] On ophthalmological evaluation, papilloedema was present in this child and there was proptosis and hypertelorism.
Management of Crouzon syndrome requires a multi-disciplinary approach and early diagnosis is very important. In the first year of life, it is preferred to release the synostotic sutures of the skull to allow adequate brain development. Repeat surgery may be needed to obtain better results.[10] If necessary, midfacial advancement and jaw surgery can be done to provide adequate orbital volume and reduce the exophthalmoses to correct the occlusion to an appropriate functional position and to provide for a more normal appearance. Prognosis usually depends on severity of the deformity.[10]
So, after receiving medical management, this child was treated by strip craniectomy. In the very young patient release of the fused suture and ensuring brain growth allows the skull to be remodeled. We have discussed with neurosurgeons further management plans. Their advice was to do nothing further. No need to give further support to the skull. Bone will grow in their natural process along the coronal plane. Because of the early diagnosis and intervention in this case, we expect a normal or near normal brain growth in this child. No complications occurred related to surgery so far.
Despite tremendous advances in the establishment of the inheritance mode of Crouzon syndrome and its prevention and treatment, it remains a significant cause of morbidity worldwide. Innovations in craniofacial surgery have enabled patients to achieve their full potential by maximizing their opportunities for intellectual growth, physical competence, and social acceptance. The importance of surgical intervention is sometimes overlooked by physicians. The idea of publishing this case report is to highlight the importance of surgical management along with that of the medical management. A timely surgical intervention can prevent major disability in adulthood. Furthermore, in expert hands, complications are rare.
Declarations
AcknowledgmentsI express my sincere gratitude to my patient and parents for their nice cooperation in spite of their sufferings.
Authors’ contributionsMain frame work of the paper: J. Morshed
Manuscript correction: M. Mutanabbi
Preparation of the manuscript: M.G. Hafiz
Financial support and sponsorshipThe first author bore all the expenses. There was neither sponsorship nor financial support.
Conflicts of interestThere are no conflicts of interest regarding publication of the paper.
Patient consentConsent was taken and strictly maintained confidentiality.
Ethics approvalAs the parents have given the consent so we did not go for ethical clearance.
REFERENCES
1. Crouzon O. Dysostose cranio-faciale héréditaire. Bulletin de la Société des Médecins des Hôpitaux de Paris 92;33:545-55. Available from: http://www.biusante.parisdescartes.fr/histoire/medica/resultats/index.php?p=2&cote=epo092&do=page. [Last accessed on Aug 24, 207].
2. Snyder-Warwick AK, Perlyn CA, Pan J, Yu K, Zhang L, Ornitz DM. Analysis of a gain-of-function FGFR2 Crouzon mutation provides evidence of loss of function activity in the etiology of cleft palate. Proc Natl Acad Sci U S A 2010;107:2515-20.
3. Fenwick AL, Goos JA, Rankin J, Lord H, Lester T, Hoogeboom AJ, van den Ouweland AM, Wall SA, Mathijssen IM, Wilkie AO. Apparently synonymous substitutions in FGFR2 affect splicing and result in mild Crouzon syndrome. BMC Med Genet 2014;15:95.
6. Gray TL, Casey T, Selva D, Anderson PJ, David DJ. Ophthalmic sequelae of Crouzon syndrome. Ophthalmology 2005;112:1129-34.
7. Gorlin RJ, Cohen MM, Levin LS. Syndromes of the head and neck. 3rd ed. Oxford: Oxford University Press; 1990. pp. 516-26.
8. Jarund M, Lauritzen C. Craniofacial dysostosis: airway obstruction and craniofacial surgery. Scand J Plast Reconstr Surg Hand Surg 1996;30:275-9.
9. Vivek P, Hegde AM, Rai K. Crouzon's syndrome: a review of literature and case report. Contemp Clin Dent 2011;2:211-4.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Morshed J, Mutanabbi M, Hafiz MG. Crouzon syndrome: an experience of surgical intervention at Bangabandhu Sheikh Mujib Medical University, Bangladesh. J Unexplored Med Data 2017;2:48-51. http://dx.doi.org/10.20517/2572-8180.2017.05
AMA Style
Morshed J, Mutanabbi M, Hafiz MG. Crouzon syndrome: an experience of surgical intervention at Bangabandhu Sheikh Mujib Medical University, Bangladesh. Journal of Unexplored Medical Data. 2017; 2: 48-51. http://dx.doi.org/10.20517/2572-8180.2017.05
Chicago/Turabian Style
Morshed, Jesmeen, Mahbub Mutanabbi, Md. Golam Hafiz. 2017. "Crouzon syndrome: an experience of surgical intervention at Bangabandhu Sheikh Mujib Medical University, Bangladesh" Journal of Unexplored Medical Data. 2: 48-51. http://dx.doi.org/10.20517/2572-8180.2017.05
ACS Style
Morshed, J.; Mutanabbi M.; Hafiz MG. Crouzon syndrome: an experience of surgical intervention at Bangabandhu Sheikh Mujib Medical University, Bangladesh. J. Unexplored. Med. Data. 2017, 2, 48-51. http://dx.doi.org/10.20517/2572-8180.2017.05
About This Article
Copyright

Author Biographies

Data & Comments
Data
0

 Cite This Article 7 clicks
Cite This Article 7 clicks

 Like This Article 0
likes
Like This Article 0
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.